10歲的小晴在出生後4個月大時,被發現不會翻身及抬頭,經確診為先天性罕病SMA第1型。1歲時因感染肺炎導致呼吸衰竭,被醫師判定生命僅剩數月,但家屬不放棄治療,4歲多開始用藥後,居家呼吸器縮減為每天使用10小時,抽痰1至2次,她從原本無法講完整句子到現在能唱完一首兒歌。
先天性罕病脊髓性肌肉萎縮症(SMA)病友、人權律師陳俊翰於今年年初突然病逝,生前嘆「有藥可用但不包括我」,引發社會大眾關注,生前他積極爭取SMA藥物治療的全面給付,促成衛福部承諾在今年「讓所有SMA病友皆有藥可用」。
今年8月1日起,健保正式擴大SMA藥物給付,並解除「3歲以下確診」與「上肢運動功能RULM≧15分」等限制,使病友免負擔每年新台幣660萬的天價藥費,預計讓原本受給付條件限制的約250位病友皆有機會獲得治療,不再落入有藥卻用不到的窘境。
被稱作「台灣SMA之父」的高雄醫學大學附設中和醫院小兒神經科教授鐘育志表示,SMA是一種單一SMN1基因變異,導致神經肌肉退化的罕見疾病。SMA症狀包括肌肉張力低下、四肢與軀幹肌肉無力與肌肉萎縮,且運動功能隨著年齡增長而持續退化。
鐘育志指出,SMA依照發病年齡和可達到的最佳運動功能,可分為4型,「約6成SMA病友在出生6個月內發病,即最嚴重的第1型,2歲前死亡率高達8成。」
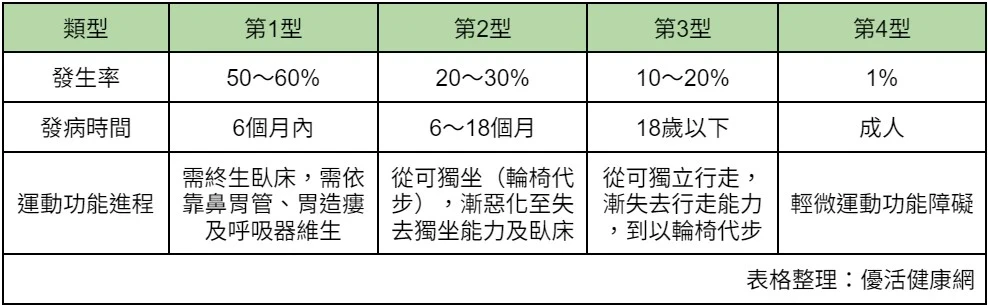
新制2大類SMA病友受惠
中國醫藥大學兒童醫院副院長周宜卿表示,台灣自2020年起分階段健保給付SMA藥物,先開放新生兒與兒童申請,隔年成為全球第1個實施全國性SMA新生兒篩檢且提供確診嬰兒藥物治療的國家,於去年健保給付3種SMA藥物,今年新制更打破確診年齡與上肢運動功能分數的限制,只要是SMA病友不分條件都有機會接受治療。
周宜卿說明,8月新制後,擴增至18歲以下發病確診SMA病友皆可用藥,其中以原本受給付門檻限制的2大類病友最為受惠:
- 針對上肢運動功能差(RULM小於15分)者:目前功能狀態較差,已嚴重到無法自行拿筷吃飯的情況,但根據文獻,接受治療也有機會改變疾病進程。
- 3歲以後發病、疾病惡化速度相對較慢者:及早積極治療,可維持或改變現有運動功能。
周宜卿指出,雖然SMA曾經無藥可醫,但隨著醫藥進展,SMA藥物不但能「幫病程踩煞車」,真實世界的臨床觀察甚至發現有機會讓運動功能進步,不論是新生兒、嬰幼兒、青少年或是成人病患,都可以與醫師討論用藥選擇,鼓勵病友再給自己一次機會,主動回診諮詢與治療。

發病年齡是疾病判別關鍵
台大醫院神經部暨腦中風中心主治醫師蔡力凱表示,從2000年台灣研究團隊研發出全球第1個SMA小鼠模式,奠定SMA致病機轉與藥物研發開始,台大醫院團隊也以此模式,在國際發表多篇重要學術論文,隨後各國陸續研發更多SMA藥物,最後自2017年起,開始提供SMA藥物跨科別照護的恩慈治療。
蔡力凱強調,許多人常將SMA誤認為漸凍症,兩者都是運動神經元疾病,但發病時間與影響不相同。漸凍症原名為「肌萎縮側索硬化症」(ALS),通常在成人時期發病,餘命可能維持3至5年;SMA則多在嬰幼兒時期發病,對家庭與社會的影響更加漫長,可能長達30、40年之久。
蔡力凱指出,受惠於醫療進展與健保罕病給付,SMA治療可以幫助病友維持神經功能,越來越多罕病病友有機會長大成年。台大醫院治療經驗顯示,接受治療的SMA病人,大多數顯示運動功能進步,且越早治療的效果越好,希望SMA病友和醫療團隊討論,並積極接受治療。(Uho優活健康網)
'%3e%3cpath%20d='M195.06%2051.1096L116.493%2038.7498C113.602%2038.2951%20110.821%2040.0467%20109.983%2042.848L99.4573%2078.0057L98.5717%2080.9652L94.4043%2094.8867L112.554%2091.5437L177.269%20112.754C180.395%20113.778%20183.75%20112.025%20184.693%20108.875L199.755%2058.5646C200.771%2055.1722%20198.561%2051.6612%20195.062%2051.1116L195.06%2051.1096Z'%20fill='%23B0DFFF'/%3e%3cpath%20d='M166.485%2077.1477C165.793%2079.4607%20167.106%2081.8963%20169.419%2082.5882C171.732%2083.2802%20174.167%2081.9675%20174.859%2079.6545C175.551%2077.3415%20174.238%2074.9059%20171.925%2074.2139C169.612%2073.522%20167.177%2074.8347%20166.485%2077.1477Z'%20fill='white'/%3e%3cpath%20d='M145.899%2070.9836C145.208%2073.2967%20146.52%2075.7322%20148.833%2076.4242C151.146%2077.1161%20153.582%2075.8034%20154.274%2073.4904C154.966%2071.1774%20153.653%2068.7418%20151.34%2068.0499C149.027%2067.358%20146.591%2068.6706%20145.899%2070.9836Z'%20fill='white'/%3e%3cpath%20d='M125.312%2064.8196C124.62%2067.1326%20125.932%2069.5682%20128.245%2070.2601C130.558%2070.952%20132.994%2069.6393%20133.686%2067.3263C134.378%2065.0133%20133.065%2062.5777%20130.752%2061.8858C128.439%2061.1939%20126.004%2062.5066%20125.312%2064.8196Z'%20fill='white'/%3e%3cpath%20d='M5.25804%200.00279501L74.4703%202.27626C77.0166%202.35929%2079.1082%204.3125%2079.3672%206.84692L82.6212%2038.6359L82.896%2041.3107L84.185%2053.8978L69.2097%2048.0718L10.3981%2055.6296C7.55918%2055.9954%204.97534%2053.9473%204.68473%2051.0985L0.0270836%205.61134C-0.287248%202.54315%202.17601%20-0.0980284%205.25804%200.00279501Z'%20fill='%230073B8'/%3e%3cpath%20d='M25.4393%2026.9418C25.6528%2029.0334%2024.1325%2030.9016%2022.041%2031.1151C19.9494%2031.3286%2018.0812%2029.8083%2017.8677%2027.7167C17.6541%2025.6251%2019.1744%2023.7569%2021.266%2023.5434C23.3576%2023.3299%2025.2258%2024.8502%2025.4393%2026.9418Z'%20fill='white'/%3e%3cpath%20d='M44.0521%2025.036C44.2656%2027.1276%2042.7453%2028.9958%2040.6537%2029.2093C38.5622%2029.4228%2036.694%2027.9026%2036.4804%2025.811C36.2669%2023.7194%2037.7872%2021.8512%2039.8788%2021.6377C41.9704%2021.4242%2043.8386%2022.9444%2044.0521%2025.036Z'%20fill='white'/%3e%3cpath%20d='M62.6668%2023.1303C62.8803%2025.2218%2061.3601%2027.09%2059.2685%2027.3036C57.1769%2027.5171%2055.3087%2025.9968%2055.0952%2023.9052C54.8817%2021.8136%2056.4019%2019.9454%2058.4935%2019.7319C60.5851%2019.5184%2062.4533%2021.0387%2062.6668%2023.1303Z'%20fill='white'/%3e%3c/g%3e%3cdefs%3e%3cclipPath%20id='clip0_926_14682'%3e%3crect%20width='200'%20height='113.047'%20fill='white'/%3e%3c/clipPath%3e%3c/defs%3e%3c/svg%3e) 無任何留言,趕緊搶頭香!
無任何留言,趕緊搶頭香!